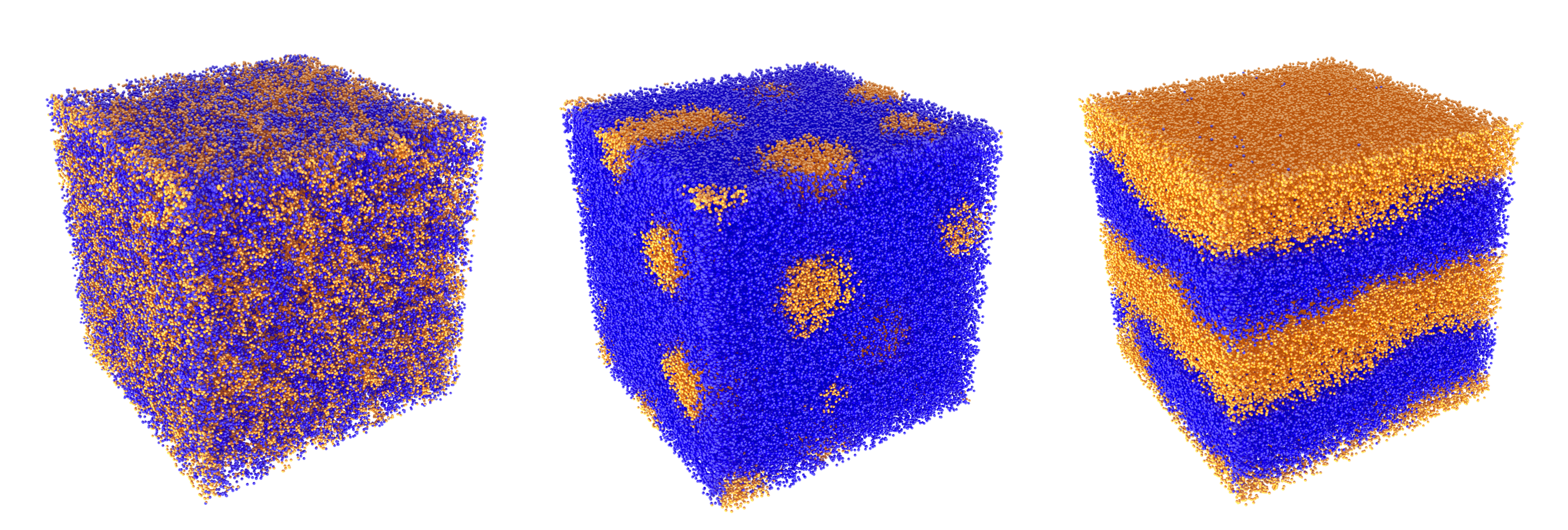
Research
We use simulation and theory to understand how polymer materials can be designed at the molecular level. Our interests include both the equilibrium and dynamic behavior of polymer materials. In particular, we study the impact of molecular features such as charge, molecular stiffness, and architecture on material properties. We also consider the impact of out-of-equilibrium forces such as strong flows on these same materials. Close collaboration with experimentalists allows us to refine our models and transfer these systems from robust theory into real applications.
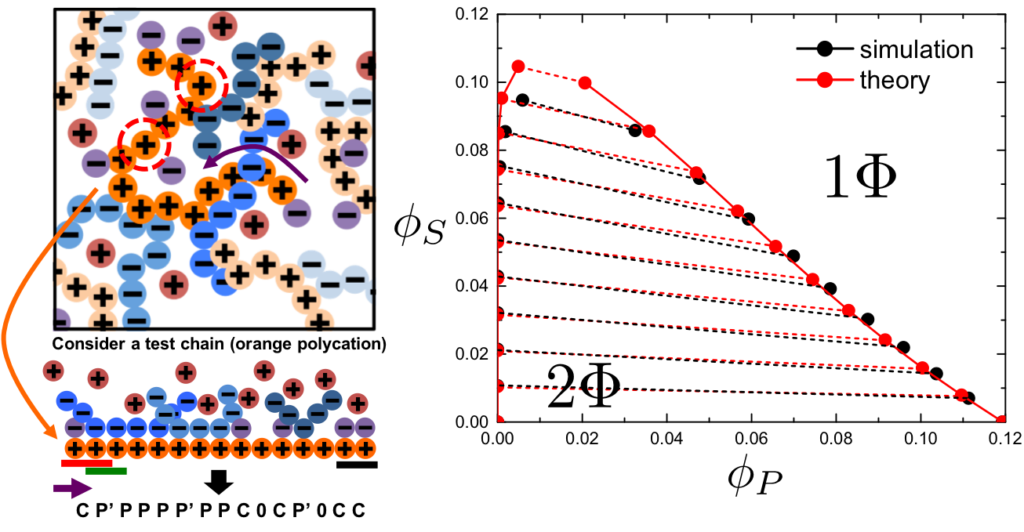
Charged polymers are used in a wide array of applications, ranging from viscosity modification and encapsulation in food products to advanced stimuli-responsive materials. Charged polymers are also ubiquitous in biological materials – most biomacromolecules are charged, which is a crucial aspect of their function. Understanding the physical properties of these systems is thus profoundly important to developing new materials that harness the features of complex biological systems (hierarchical structure, defined monomer sequences, etc.) for the wide array of applications that already feature charged polymers (and more!).
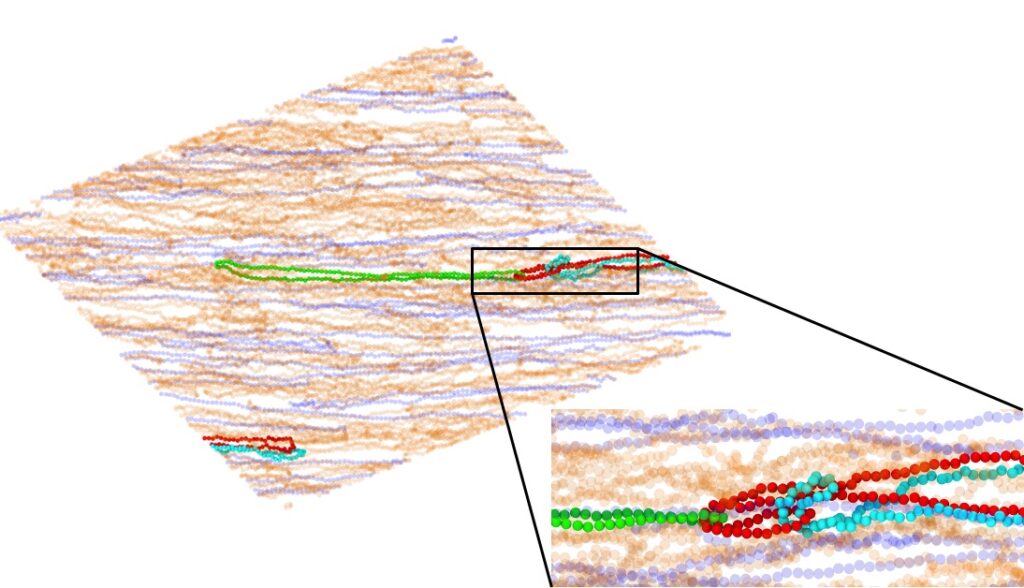
We are developing methods to simulate semidilute polymer solutions that are driven strongly out of equilibrium, which is common in polymer processing flows. Importantly, we resolve how the molecular conformations of polymers are affected by flow in the presence of chain-chain interactions and hydrodynamics.
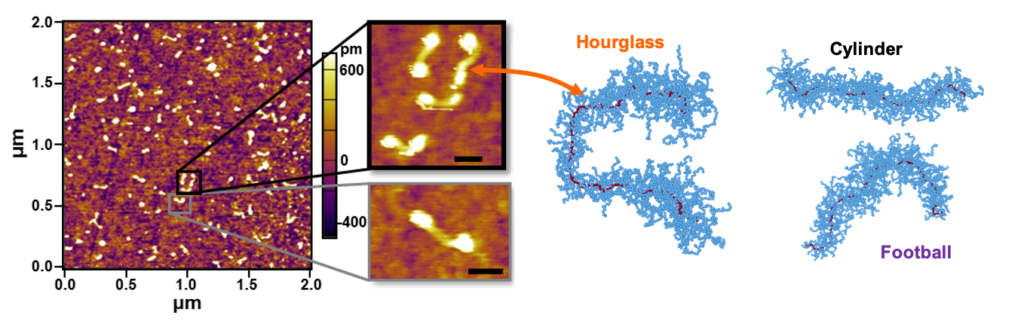
Bottlebrush polymers are branched polymers characterized by densely-grafted side-chains, which have a large number of prospective applications in advanced materials. These polymers can be used to form photonic crystals with ‘structural color’, behave as pressure-sensitive adhesives, and form low-modulus elastomers. The usefulness of these polymers is based on their unique properties – the densely-grafted side chains increase molecular stiffness, suppresses entanglements, and imbue the polymer with a molecular ‘thickness’ that provides another tunable molecular parameter for molecular design.